Alpha-1 antitrypsin deficiency, or Alpha-1, is one of the most common serious genetic conditions worldwide. Alpha-1 can cause severe lung and/or liver disease and is a leading reason for lung transplantation in adults and liver transplantation in young children. An estimated 100,000 Americans have the condition, yet only ten percent have been accurately diagnosed. Those with Alpha-1 call themselves “Alphas.”
What is alpha-1 antitrypsin?
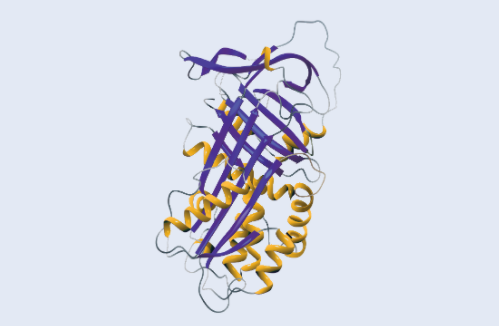
Alpha-1 antitrypsin or AAT, is a protein manufactured primarily in the liver. You may sometimes see it referred to as “alpha-1 proteinase inhibitor” or “aPi.” AAT’s function is to protect the tissues of the body from being damaged by proteolytic enzymes — substances that break down proteins. When the body is exposed to agents such as bacteria and viruses, the inflammatory and immune systems are activated. This sends signals to white blood cells and other cellular components to mobilize and destroy the invading agents. A type of white blood cell, the neutrophil, is one of the first-line defenders in this process, and it is important for its ability to chew up foreign invaders. Neutrophils use a variety of proteolytic enzymes in defending the body. One of these is called “neutrophil elastase.”
Normally, a balance exists in the body between AAT and neutrophil elastase. In all individuals, AAT’s normal function is to protect the tissues of the body against proteolytic enzymes, especially during inflammation.
What is alpha-1 antitrypsin deficiency?
Alpha-1 antitrypsin deficiency is not a disease, but an inherited condition that increases an individual’s risk of developing disease, particularly of the lungs and the liver. Alpha-1 is characterized by a reduced blood level of the protein alpha-1 antitrypsin ( AAT).
What causes Alpha-1?
Virtually all the alpha-1 antitrypsin in the blood is produced in the liver. In individuals with Alpha-1, the decreased levels of AAT are the result of abnormalities in the AAT gene. This abnormality causes the AAT to function poorly, to be poorly manufactured, or most commonly, to be poorly released from the liver cells.
Who has alpha-1 antitrypsin deficiency?
Although Alpha-1 is often viewed as an extremely rare condition, in reality, it is among the most common genetic conditions affecting individuals in the United States and Europe, and it is seen in virtually all populations worldwide. In the United States, it is estimated that approximately 100,000 individuals have a severe deficiency of this important protein, while more than 20 million Americans are carriers of a single abnormal AAT gene. A similar number of people is believed to be affected in Europe. The vast majority of individuals with Alpha-1 and carriers of a single abnormal AAT gene remain undiagnosed throughout the world.
What causes disease in Alphas?
Since alpha-1 antitrypsin’s normal function is to protect the tissues of the body against proteolytic enzymes, this reduced blood level is what accounts for the increased susceptibility to certain diseases in individuals with Alpha-1.
The lungs and liver are most well known to be at risk for damage among individuals with Alpha-1. People who develop disease are often diagnosed with emphysema, a destructive disease of the air sacs of the lung, or cirrhosis, liver cell injury with scarring. Necrotizing panniculitis, an inflammatory condition of the skin, and vasculitis, an inflammation of the blood vessels, are other conditions that have also been associated with Alpha-1. Injury to the lungs, skin, and blood vessels appears to be related to the destruction of a protein known as “elastin.”
Many of the risk factors that can lead to disease in Alphas are known, and the management of these risk factors is one of the most important ways in which individuals with Alpha-1 can directly affect the quality of their lives. Recently, there has been a growing appreciation that individuals who have been identified as carriers of the Alpha-1 gene also may be at increased risk for disease development. Therefore, limiting exposure to environmental risk factors is important for these individuals as well.
How is Alpha-1 diagnosed?
There are currently three tests available that can help determine whether or not an individual is an Alpha or Alpha-1 carrier which include serum level, phenotyping, and genotyping. Experts recommend that at least two tests be conducted in order to verify the diagnosis of alpha-1 antitrypsin deficiency.
Why is Alpha-1 so underdiagnosed?
Studies have shown that, once a patient with Alpha-1 develops symptoms, it takes an average of seven years and visits to three to five different doctors before the diagnosis of Alpha-1 is made. Because diseases associated with Alpha-1, such as emphysema and cirrhosis of the liver also occur in those without Alpha-1 and can sometimes be attributed to lifestyle choices like cigarette smoking and alcohol consumption, the diagnosis of Alpha-1 is frequently overlooked.
Who should be tested for Alpha-1?
Until it is appropriate to begin screening the general population for Alpha-1, there are some guidelines as to who should be tested for this condition. These include individuals with:
- A diagnosis of emphysema, chronic bronchitis, unexplained bronchiectasis, or chronic obstructive pulmonary disease (COPD)
- Family history of Alpha-1
- Family history of emphysema, bronchiectasis, liver disease, or panniculitis
- Chronic asthma in which lung function does not return to normal with therapy in adolescents and adults
- Recurrent pneumonia or bronchitis
- Unexplained liver disease
- Granulomatosis with polyangiitis (GPA or C-ANCA positive vasculitis)
- Necrotizing panniculitis